USP18 modulates the sensing of HIV‑1
Project Member
")
Klinik für Gastroenterologie,
Hepatologie und Infektiologie
Universitätsklinikum Düsseldorf
MNR-Klinik / Gebäude: 13.54
Moorenstrasse 5
40225 Düsseldorf
Chaohui Lin
Project Summary
The RNA and cDNA of HIV‑1 (human immunodeficiency virus type 1) can be recognized and sensed by macrophage and dendritic cell-expressed endosomal Toll-like receptors and cytosolic sensor, cGAS respectively. cGAS-cDNA interaction induces cGAMP production, which stimulates STING dimerization and activation. STING mediates TBK1 activation, which subsequently phosphorylates IRF3, driving IFN‑α/β induction. IFN‑α/β signals back via the IFN‑α receptor 1 and 2 (IFNAR1/2), inducing a plethora of IFN stimulated gene (ISGs), including ISG15 and p21. p21 exhibits antiviral activity against retroviruses by activating SAMHD1 restriction function and repressing key enzymes involved in de novo dNTP biosynthesis. ISG15 exhibits antiviral activity by activating cellular factors required to block viral infection through ISGylation.
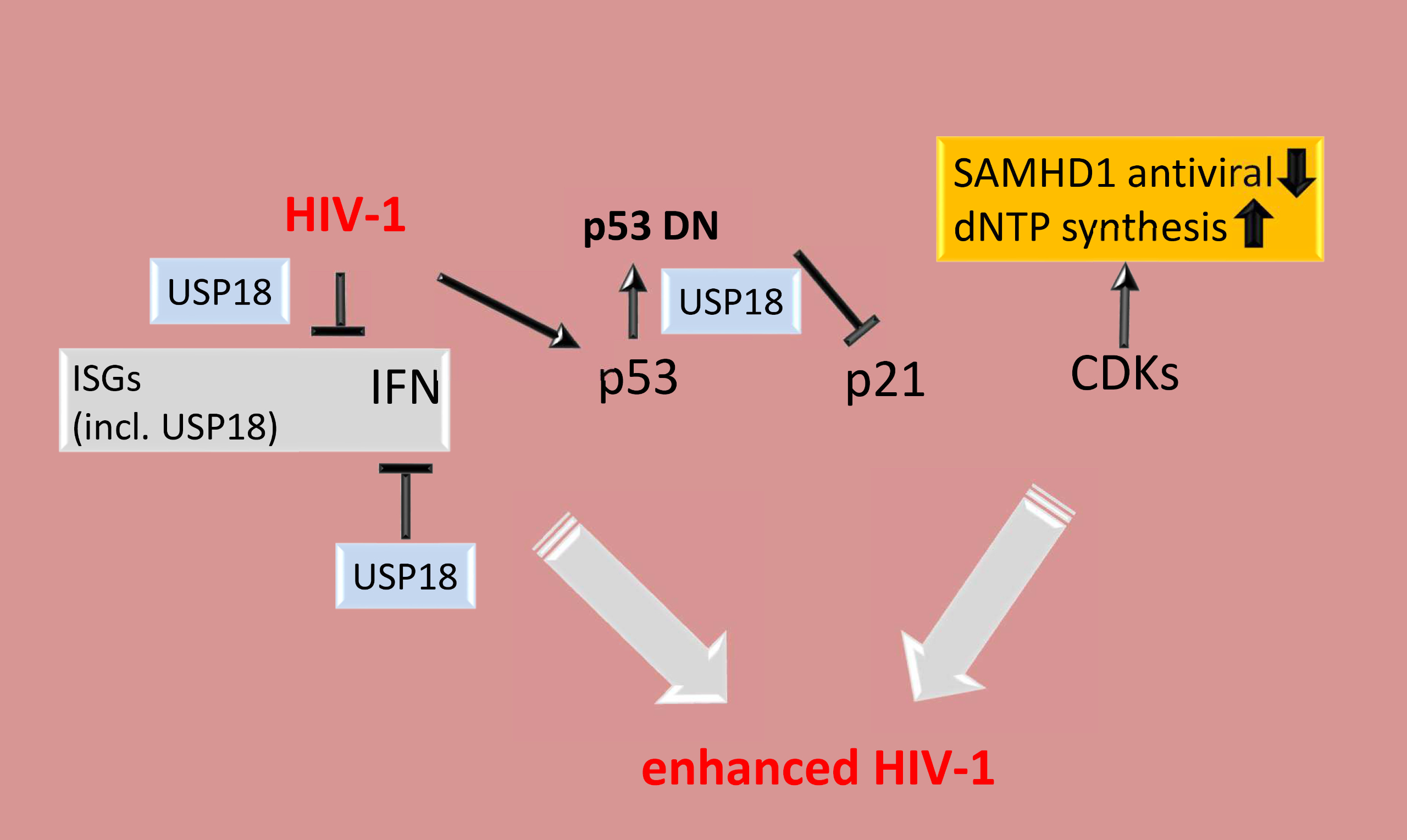
USP18 is an ISG15-specific isopeptidase, and negatively regulates IFN signaling. USP18 also abrogates NF-κB signaling by mediating TAK1 and NEMO deubiquitination in a protease dependent and independent manner. Although USP18 plays an important role in innate immune responses by negatively regulating type I and III IFNs and mediates deISGylation of key antiviral proteins, its role in HIV‑1 infection, recognition and sensing in innate target cells has not been explored.
Within the framework of SPP1923, we showed in our first publication that HIV‑1 induces USP18 expression and the presence of USP18 contributes to HIV‑1 replication by abrogating p21 antiviral function. In a follow-up work, we provided a mechanistic detail to USP18-mediated abrogation of p21 antiviral activity. We confirmed that p53 is HIV-1-inducible. However, USP18, by its protease activity, accumulated misfolded dominant negative p53, which abrogated wild type p53 transactivation of p21 leading to an expanded intracellular dNTP pool, inactivated SAMHD1 and increased HIV‑1 reverse transcription products.